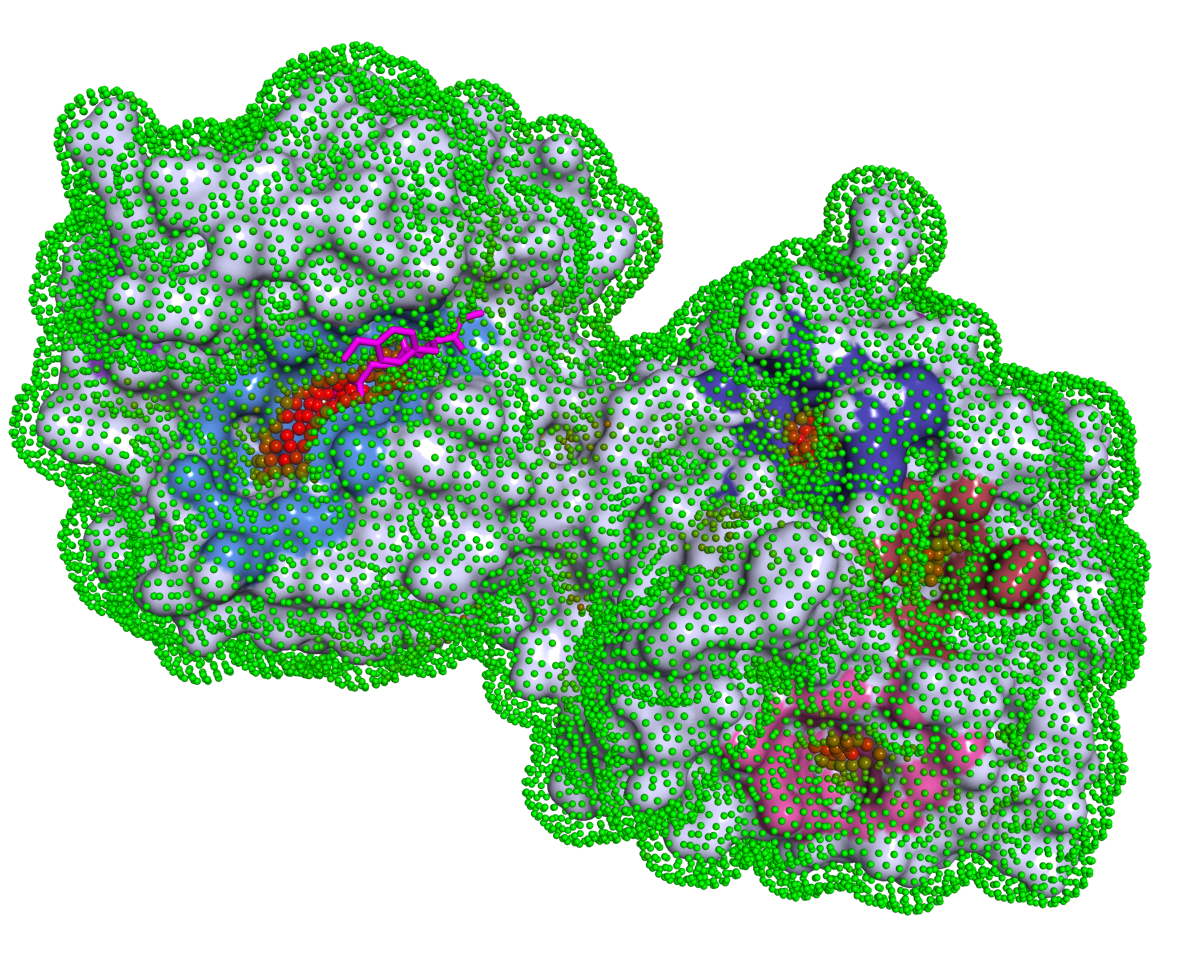
Introduction
Slides with method overview: p2rank_slides
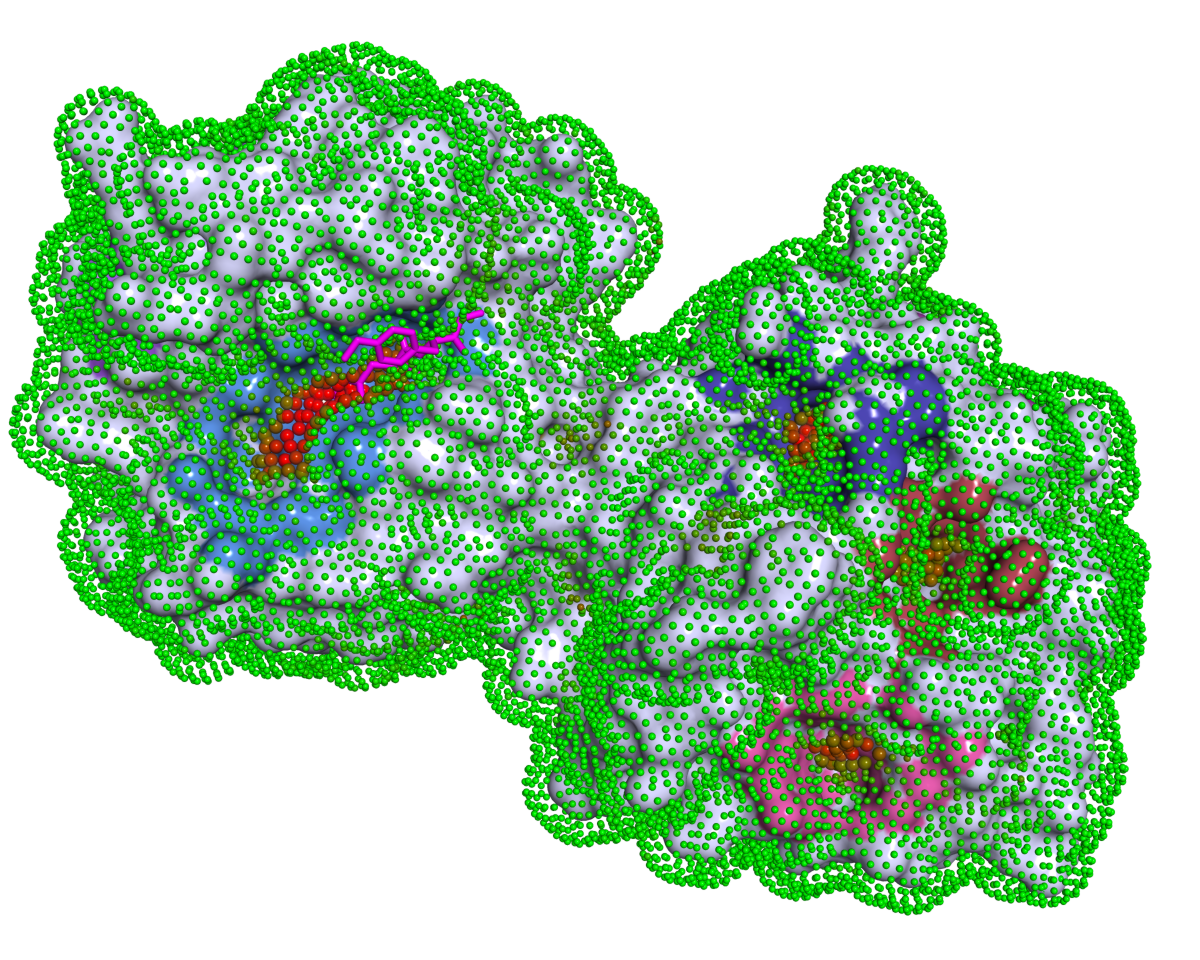
Download
Stand-alone, platform independent binary packages are available here:
See also github.com/rdk/p2rank for the latest README instructions.
Source Code
Source code and documentation is available on GitHub: github.com/rdk/p2rank
Web Version
Web interface for the method is called PrankWeb and is now available at prankweb.cz
PDBe-KB
Bidning site predictions for large part of PDB are now available via PDBe Knowledge Base at www.ebi.ac.uk/pdbe/pdbe-kb . You can find P2Rank predictions among Functional anotations / Predicted ligend binding sites, see for example www.ebi.ac.uk/pdbe/pdbe-kb/proteins/2etx#annotations .
Publications
If you use P2Rank, please cite relevant papers:
- Software article in JChem about P2Rank pocket prediction tool: Krivák R, Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. Journal of Cheminformatics. 2018 Aug.
- Conference paper inroducing P2Rank prediction algorithm: Krivák R, Hoksza D. P2RANK: Knowledge-Based Ligand Binding Site Prediction Using Aggregated Local Features. International Conference on Algorithms for Computational Biology 2015 Aug 4 (pp. 41-52). Springer, Cham.
- Research article in JChem about PRANK rescoring algorithm: Krivák R, Hoksza D. Improving protein-ligand binding site prediction accuracy by classification of inner pocket points using local features. Journal of Cheminformatics. 2015 Dec;7(1):12.
- Web-server article in NAR: Jendele L, Krivak R, Skoda P, Novotny M, Hoksza D. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Research, Volume 47, Issue W1, 02 July 2019, Pages W345–W349
Feedback
We would be happy to hear about your use cases, experiences and ideas/feature requests. Either raise an issue on GitHub issue tracker or get in touch by mail. Please address any correspondence to both (rkrivak [at] gmail.com) and (david.hoksza [at] gmail.com).
About
Developed at Charles University Structural Bioinformatics Group.