Bioinformatics & Cheminformatics
 MolArt
(MolArt is a responsive, easy-to-use JavaScript plugin which enables users to view annotated protein sequence and overlay the annotations over a corresponding experimental or predicted protein structure.)
MolArt
(MolArt is a responsive, easy-to-use JavaScript plugin which enables users to view annotated protein sequence and overlay the annotations over a corresponding experimental or predicted protein structure.)
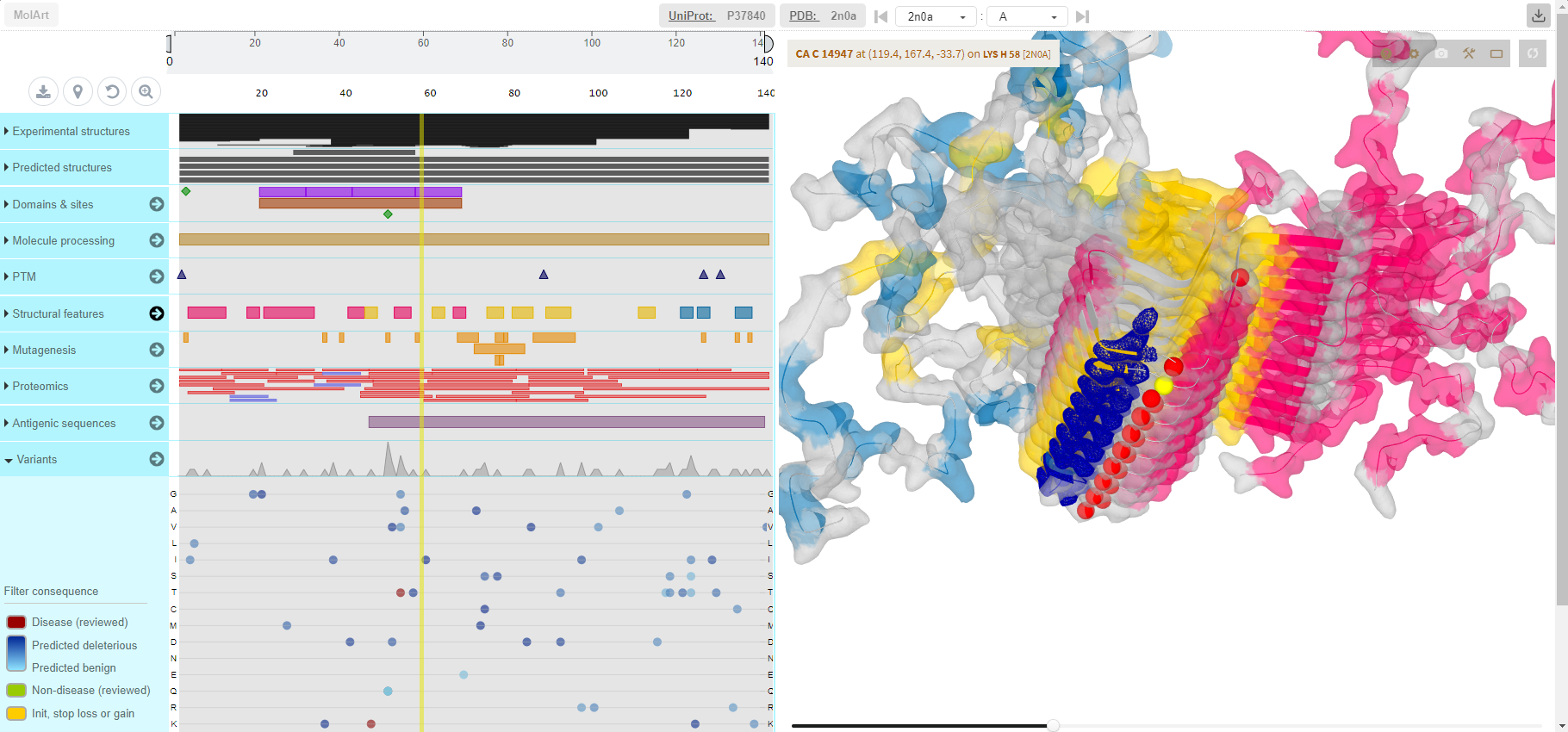
MolArt is a responsive, easy-to-use JavaScript plugin which enables users to view annotated protein sequence (including variation data from large scale studies) and overlay the annotations over a corresponding experimental or predicted protein structure. It couples protein sequence annotation capabilities provided by ProtVista (or more precisely its modified responsive version implemented when developing MolArt) with structure visualization capabilities provided by LiteMol. Since it does not have any software dependencies and all the data are obtained on the fly, it is easy to integrate it to any web page. |
|
Molpher aims to be a scalable and interactive software tool to aid exploration of chemical space, the vast universe containing all possible compounds. Many areas of chemical biology, such as drug discovery, rely heavily on chemical libraries offering compounds usable in the industrial processes. Given a set of molecules with desired characteristics, Molpher explores their common neighbourhood based on structural similarity, as it represents promising part of the chemical space to find new additions into those libraries. In order to decrease the chance of missing interesting parts of the space, Molpher offers the human researcher to observe and interactively alter the exploration process. Generated subspace is expected to be further tested for synthesizability and biological activity by other software tools. Among main features, Molpher offers optimized parallel exploration algorithm, compound logging, dimension-reduced visualization of chemical space and interactive widget-based GUI. Codebase is extensible in terms of additional morphing operators, chemical fingerprints, similarity measures and visualization strategies to allow further experiments. |
 P2Rank
(Protein binding site prediction)
P2Rank
(Protein binding site prediction)
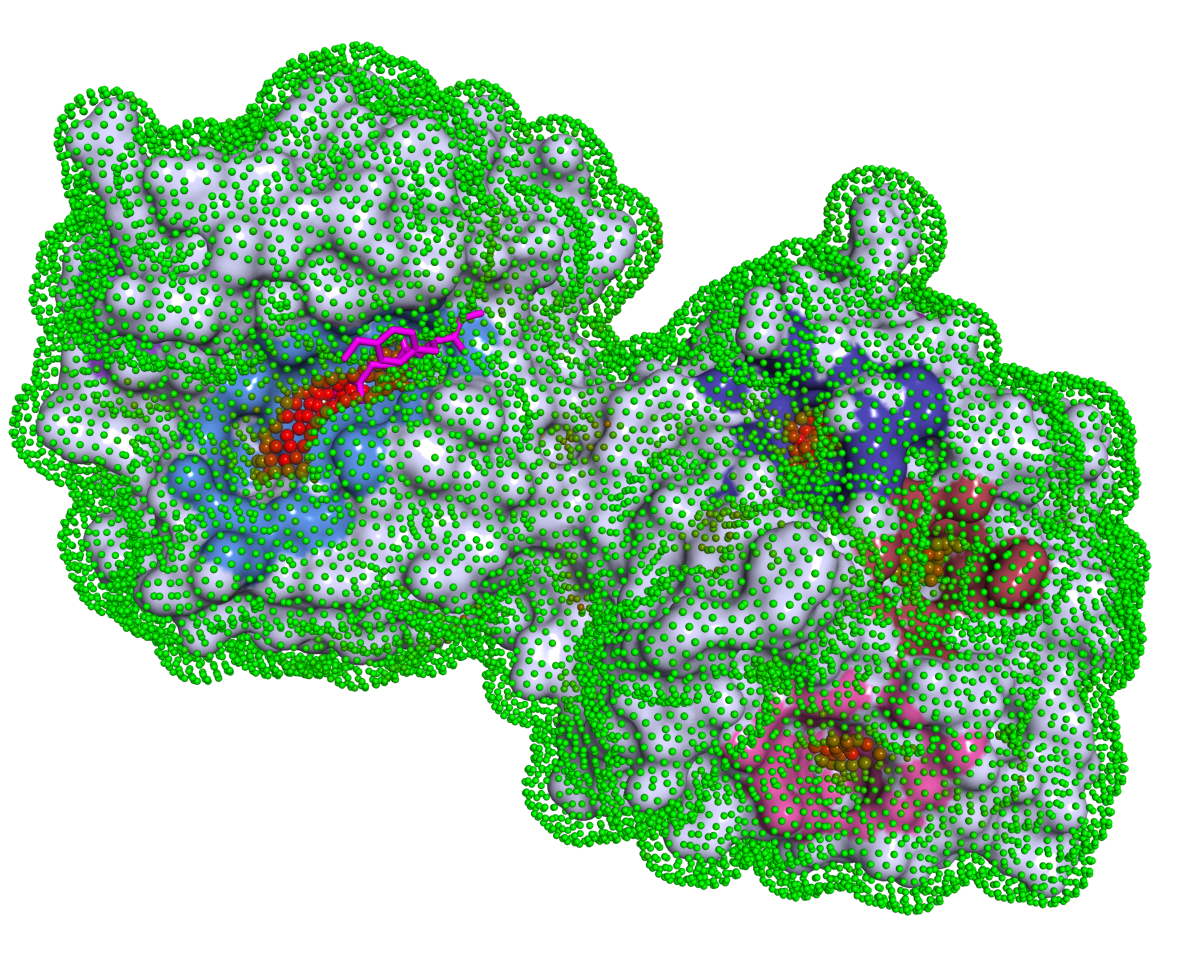
P2Rank is a machine learning based method for prediction of binding sites from protein structure. P2Rank uses Random Forests classifier to infer ligandability of local chemical neighborhoods near the protein surface which are represented by specific near-surface points and described by aggregating physico-chemical features projected on those points from neighboring protein atoms. The points with high predicted ligandability are clustered and ranked to obtain the resulting list of binding site predictions. P2Rank is freely available at https://prankweb.cz/.
|
 rPredictor
(Infrastructure consisting of tools, a database and web interface that together enable predicting ribosomal RNA secondary structure and analysing it.)
rPredictor
(Infrastructure consisting of tools, a database and web interface that together enable predicting ribosomal RNA secondary structure and analysing it.)
rPredictor is a bioinformatical infrastructure consisting of tools, a database and a web interface that together enable predicting ribosomal RNA secondary structure and analysing it. The predicted structures, their analyses and further details about the rRNA molecules are accessible through this website.
The aim of rPredictor is to develop and deploy a technique of predicting ribosomal RNA secondary structure and make the resulting structural information readily available. At the same time, the rPredictor database contains rich annotations of rRNA structures and the underlying sequences.
The project is being developed at the Faculty of Mathematics and Physics, Charles University in Prague in close cooperation with the bioinformatics laboratory of Microbiology Institute AVČR. |
 SETTER
(RNA structure similarity search)
SETTER
(RNA structure similarity search)
SETTER web-server utilizes SETTER (SEcondary sTructure-based TERtiary Structure Similarity Algorithm) method for fast and accurate structural pairwise alignment. The server is capable of comparing a pair of RNA structures or using one strucutre as a query and search against a user-defined database of RNA structures. The efficiency of the algorithm is given by the decomposition of the RNA structure into the set of non-overlapping generalized secondary structure motifs (GSSUs). GSSU usually resembles a hairpin motif possibly containing bulges and/or internal loops in its stem part. A segmentation to GSSUs offers good scalability with respect to the structure size (SETTER scales linearly with the structure size) because the number of residues in GSSUs (SETTER scales quadratically with the GSSU size) generally does not increase with increased size of the RNA structure. The underlying SETTER algorithm is both accurate and very fast, and does not impose limits on the size of aligned RNA structures. SETTER is able to compare a pair of even the largest RNA structure in less than one minute. |
 Traveler
(Traveler is an RNA sescondary structure visualization tool implementing a template-based approach enabling to lay out even the largest RNA structures in the standard orientation.)
Traveler
(Traveler is an RNA sescondary structure visualization tool implementing a template-based approach enabling to lay out even the largest RNA structures in the standard orientation.)
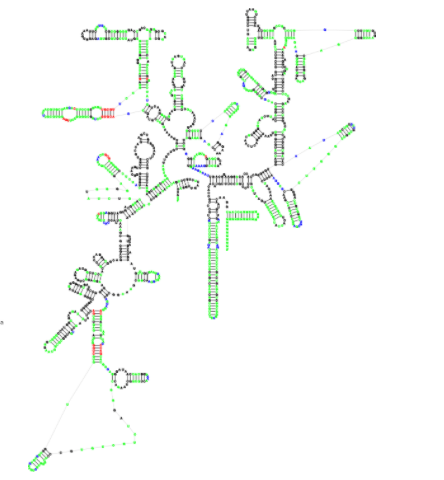
Visualization of RNA secondary structures is a complex task, and, especially in the case of large RNA structures where the expected layout is largely habitual, the existing visualization tools often fail to produce suitable visualizations. This led us to the idea to use existing layouts as templates for the visualization of new RNAs similarly to how templates are used in homology-based structure prediction. Traveler is a software tool enabling visualization of a target RNA secondary structure using an existing layout of a sufficiently similar RNA structure as a template. Traveler is based on an algorithm which converts the target and template structures into corresponding tree representations and utilizes tree edit distance coupled with layout modification operations to transform the template layout into the target one. Traveler thus accepts a pair of secondary structures and a template layout and outputs a layout for the target structure. Traveler is used by RNACentral for visualization of all the secondary structures available in RNACentral.
|
General Indexing
|
PGRTree
(Plugin for Indexing Multidimensional Data in PostgreSQL Using R-tree)
In commercial database platforms, the standard search over multiple attributes is provided by B+-tree (or it’s variants) with compound keys. On the other hand, such systems provide also multidimensional indexing, however, just for spatial purposes (such as GIS or CAD applications) and use special data types and querying syntax. Our solution allows to apply R-tree index (hence multi-dimensional indexing structure) in the same way as B+-tree with compound keys is applied. The solution is delivered as a plugin for PostgreSQL database. |